Classical and quantum computing reveal molecular insights
To study molecules with complex structures, UChicago and IBM combined forces to develop a new method that takes advantage of today’s quantum computers.
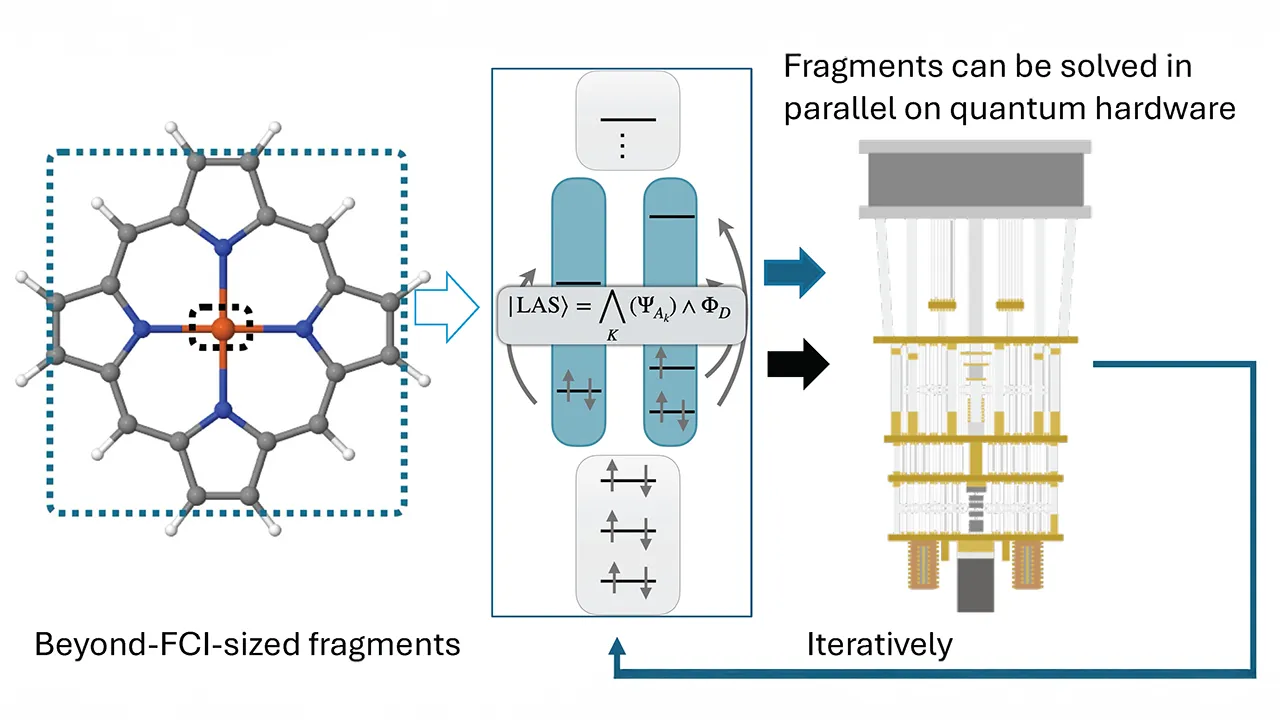
A new method developed by the University of Chicago and IBM reveals insights into the complex electronic structures of molecules. (Image courtesy of Wang et al.)
A new computational framework developed by the University of Chicago Pritzker School of Molecular Engineering (UChicago PME), the Department of Chemistry and IBM combines the power of classical and quantum computing to reveal insights into the complex electronic structures of molecules.
Called LASSQD (localized active space sample-based quantum diagonalization), the method breaks down the longstanding challenge of accurately modeling molecules into smaller fragments, then employs a quantum sampling technique to solve them, which in turn allows the hybrid algorithms to solve larger fragments.
Researchers showed that, although the method produces approximate solutions in the fragments, its results remain highly accurate while using a smaller dimension space and can run on current “noisy” quantum computers, which do not yet have full error-correction.
The results, published in the Proceedings of the National Academy of Sciences, could help advance computational research in areas including catalysis, energy, and chemical discovery. The work was done under the IBM-University of Chicago Quantum Collaboration.
“This work demonstrates that current quantum hardware can already be integrated with established quantum chemistry methods. Although today's devices are still limited, they can already play a useful role in hybrid computational workflows,” said Prof. Laura Gagliardi, who co-authored the research.
This work demonstrates that current quantum hardware can already be integrated with established quantum chemistry methods.
Prof. Laura Gagliardi, co-author of the research
A classical-quantum hybrid workflow to understand molecules
Understanding a molecule’s ground state— the lowest energy state—has important implications for studying chemical reaction and catalysis, which underpin much of the global chemical industry. Determining the ground state requires calculations on how a molecule’s electrons are arranged and how those electronic configurations contribute to its energy.
That’s an extremely difficult problem. Molecules like transition-metal compounds, which are highly relevant to energy and catalysts, often have several electronic configurations competing with each other. Accurately describing these systems requires solving the Schrödinger equation, which predicts the quantum-mechanical behavior of electrons. However, the computational cost of obtaining exact solutions grows exponentially.
Gagliardi and her team have developed multireference theories and methods to tackle this challenge. The local active space methods partition the molecular space into fragments, and solve the fragment Schrödinger equation self-consistently. By combining the fragment solutions, the approach makes it possible to study larger and more complex molecular systems while retaining high accuracy than without fragmentation methods.
To explore whether current quantum computers could help provide even better solutions, Gagliardi’s team collaborated with researchers at IBM.
Quantum computers are naturally suited to simulating quantum systems because they process information according to the same quantum-mechanical principles that govern electrons and molecules. But current quantum computers, called noisy intermediate-scale quantum computers, are sensitive to environmental interreference and do not yet support the fully fault-tolerant error correction required for large-scale quantum computation.
Still, the team developed a method in concert with IBM’s sample-based quantum diagonalization algorithm to treat individual molecular fragments. The quantum calculations identify the most important electronic configurations within each fragment, enabling the classical LAS method to solve substantially larger fragment problems than would otherwise be feasible. The technique began with computations on a classic computer; when the method required quantum computing, the team prepared a quantum circuit that gets sent to IBM’s quantum computer. When the results returned, the team finished the computations classically.
“The results are still approximate, but the accuracy is not compromised when you use this hybrid workflow,” said Joanna (Qiaohong) Wang, the UChicago PME graduate student who is the first author of the paper.

Harnessing the power of quantum computation
The team applied the method to several iron-containing systems, including a model of catalytic center in metal-organic frameworks (MOFs), as well as the spin gap of iron porphyrin—a complex transition metal widely used in industry. These proof-of-concept studies showed that the fragment-based calculations retained high accuracy without requiring an exact treatment of the entire molecular system.
Exact calculations may be performed on quantum computers in the future, but for now, “this work demonstrates that current quantum hardware can already be incorporated into hybrid quantum chemistry workflows. While today's devices remain limited, they can already contribute to chemically meaningful calculations when combined with robust classical methods,” said Gagliardi, who is also the Richard and Kathy Leventhal Professor in the Department of Chemistry.
Next the team is working to improve the method to reduce computational costs. The ultimate goal is to harness the power of quantum computing to generate new insights or predictions into how chemicals behave.
“Classically solving these problems still incurs huge computational costs, but quantum computing will eventually be able to solve these problems quantum-mechanically without such costs,” Wang said. “We show that we are able to use current quantum devices to get a benefit now, but future capabilities will be really exciting.”
Other authors on the paper include Kevin J. Sung, Ruhee D’Cunha, Matthew R. Hermes, Tanvi Gujarati, Yukio Kawashima, Yu-ya Ohnishi, Gavin O. Jones, and Mario Motta.
Citation: “Localized sample-based quantum diagonalization for strongly correlated chemistry,” Wang et al, Proceedings of the National Academy of Sciences, July 10, 2026. DOI: 10.1073/pnas.2603914123
Funding: IBM, Department of Energy, National Science Foundation
Related News
-
New method could yield fast, cross-country quantum network
July 09, 2024Quantum computers offer powerful ways to improve cybersecurity, communications, and data processing, among other fields. To realize these full benefits, however, multiple quantum computers need to be... -

Innovative phonon-based quantum computing research at UChicago Engineering gets a $3 million boost by the U.S. Department of Defense
July 15, 2024UChicago Pritzker School of Molecular Engineering Prof. Andrew Cleland has been named a 2024 Vannevar Bush Faculty Fellow, the U.S. Department of Defense’s flagship single-investigator award for basic... -

US Defense Department, State of Illinois announce multimillion-dollar quantum testing program
July 16, 2024The US Department of Defense will invest up to $140 million to develop a national proving ground for quantum technologies in the Chicago region, Illinois Governor JB Pritzker announced Tuesday morning...